google.com, pub-6401434982018401, DIRECT, f08c47fec0942fa0
Health Tips | Healthy Life Ideas | Expert Weight Loss | Animal Health Care | Best Parenting Tips | Diabetes Care | Digestive CareHealth Life AI
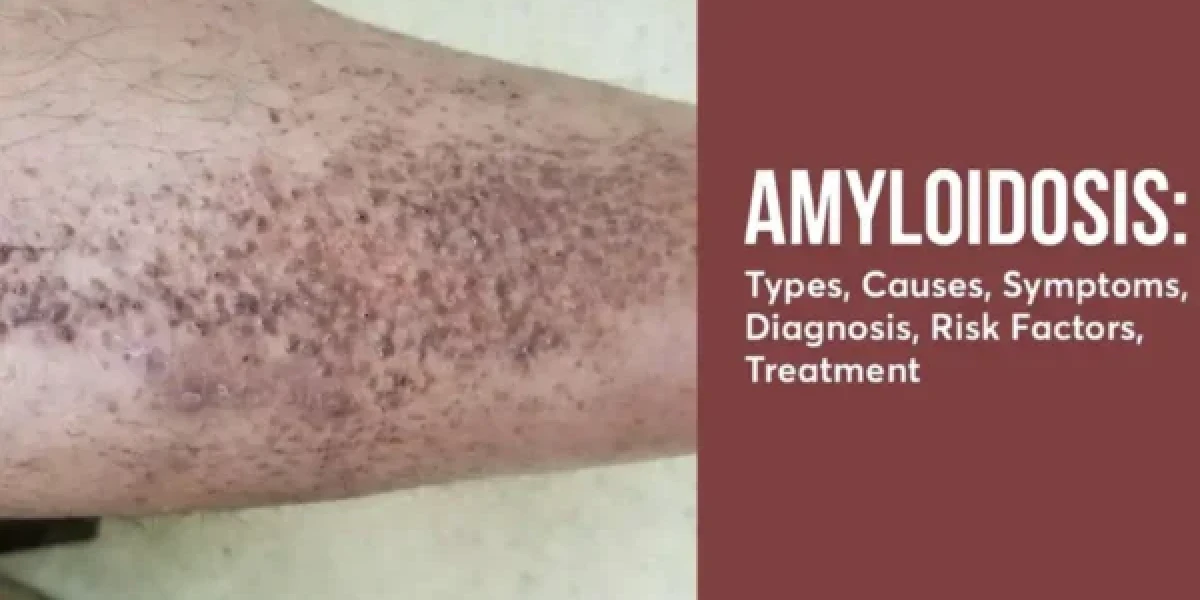
Amyloidosis is a group of rare diseases that occur when a protein called amyloid builds up in various organs and tissues of the body. Amyloid is an abnormal protein that is produced by the bone marrow or by certain cells in response to inflammation or infection. Amyloid can accumulate in any part of the body, but it commonly affects the heart, kidneys, liver, nerves, and digestive system. Amyloidosis can cause organ failure, symptoms such as fatigue, swelling, numbness, and diarrhea, and complications such as bleeding, infections, and blood clots.
Understanding amyloidosis is important because it can help with the diagnosis, treatment, and prevention of this condition. Amyloidosis is often misdiagnosed or diagnosed late because of its nonspecific and variable signs and symptoms. A timely and accurate diagnosis can improve the prognosis and quality of life of people with amyloidosis. Treatment options for amyloidosis depend on the type, cause, and extent of the disease. Some treatments aim to reduce the production or remove the source of amyloid, while others aim to manage the symptoms and complications of organ damage. Prevention strategies for amyloidosis include avoiding or treating the underlying conditions that can trigger amyloid formation, such as chronic infections, inflammatory diseases, or genetic disorders.
Causes of Amyloidosis
Amyloidosis is caused by the abnormal accumulation of amyloid proteins in various organs and tissues of the body. There are many different types of amyloidosis, each with a different cause and a different type of amyloid protein. The main causes of amyloidosis are:
Abnormal protein accumulation: This is the most common cause of amyloidosis. It occurs when the bone marrow produces abnormal antibodies or fragments of antibodies called light chains. These light chains can clump together and form amyloid fibrils, which are insoluble and deposit in various organs and tissues. This type of amyloidosis is called AL amyloidosis or primary amyloidosis. It usually affects the heart, kidneys, liver, and nerves.
Genetic factors: This is a rare cause of amyloidosis. It occurs when a person inherits a mutation in a gene that codes for a protein that can form amyloid fibrils. These proteins can be normal components of the body, such as transthyretin, which is a protein that transports thyroid hormones and vitamin A, or abnormal proteins, such as lysozyme, which is an enzyme that breaks down bacteria. This type of amyloidosis is called hereditary or familial amyloidosis. It usually affects the nerves, heart, kidneys, and eyes.
Chronic inflammatory conditions: This is another rare cause of amyloidosis. It occurs when a person has a long-term or recurrent inflammatory disease, such as rheumatoid arthritis, Crohn’s disease, or tuberculosis. These diseases can stimulate the liver to produce a protein called serum amyloid A (SAA), which is part of the acute phase response to inflammation or infection. SAA can break down and form amyloid fibrils, which can be deposited in various organs and tissues. This type of amyloidosis is called AA amyloidosis or secondary amyloidosis. It usually affects the kidneys, spleen, liver, and intestines.
Types of Amyloidosis
Amyloidosis is a group of rare diseases that occur when a protein called amyloid builds up in various organs and tissues of the body. Amyloid is an abnormal protein that is produced by the bone marrow or by certain cells in response to inflammation or infection. Amyloid can accumulate in any part of the body, but it commonly affects the heart, kidneys, liver, nerves, and digestive system.
There are many different types of amyloidosis, each with a different cause and a different type of amyloid protein. The main types of amyloidosis are:
Systemic amyloidosis: This is the most common type of amyloidosis, which affects multiple organs and tissues throughout the body. Systemic amyloidosis can be further classified into subtypes based on the source and nature of the amyloid protein. Some of the common subtypes are AL amyloidosis, AA amyloidosis, and ATTR amyloidosis.
Localized amyloidosis: This is a rare type of amyloidosis, which affects only one organ or tissue. Localized amyloidosis can be caused by overproduction of amyloid by the cells in that specific area, or by deposition of amyloid from the blood. Localized amyloidosis can affect the skin, eyes, lungs, brain, or other parts of the body.
Primary and secondary amyloidosis: These are terms that are sometimes used to describe the cause of amyloidosis. Primary amyloidosis refers to amyloidosis that occurs without any known underlying disease or trigger. Secondary amyloidosis refers to amyloidosis that occurs as a complication of another disease or condition, such as chronic infection, inflammation, or cancer
Symptoms of Amyloidosis
Amyloidosis can cause a variety of symptoms, depending on the type of amyloidosis and the organs and tissues affected. The symptoms of amyloidosis may vary from person to person and may develop gradually or suddenly. Some of the common symptoms of amyloidosis are:
Varied presentation depending on organs affected: Amyloidosis can affect any organ or tissue in the body, and the symptoms may reflect the specific organ dysfunction or damage. For example, amyloidosis that affects the heart may cause shortness of breath, chest pain, irregular heartbeat, or fluid retention. Amyloidosis that affects the kidneys may cause protein in the urine, swelling of the legs, high blood pressure, or kidney failure. Amyloidosis that affects the nerves may cause numbness, tingling, pain, or weakness in the hands or feet. Amyloidosis that affects the digestive system may cause diarrhea, constipation, nausea, vomiting, or weight loss.
Generalized symptoms: Amyloidosis can also cause some general symptoms that are not specific to any organ or tissue. These symptoms may include fatigue, weakness, loss of appetite, weight loss, fever, night sweats, or skin changes. These symptoms may be caused by the effects of amyloid on the blood vessels, the immune system, or the metabolism.
Diagnosis of Amyloidosis
Amyloidosis can be difficult to diagnose because the symptoms can be vague and similar to those of other diseases. A diagnosis of amyloidosis requires a combination of clinical evaluation, imaging studies, biopsy, and laboratory tests. Some of the methods used to diagnose amyloidosis are:
Clinical evaluation: This involves a thorough medical history and physical examination of the patient, to assess the signs and symptoms, risk factors, and family history of amyloidosis. The clinical evaluation may also include a review of the patient’s medications, as some drugs can cause or worsen amyloidosis.
Imaging studies: These are tests that use various techniques to create images of the organs and tissues affected by amyloidosis. Imaging studies can help to evaluate the extent and severity of organ damage and to monitor the response to treatment. Some of the common imaging studies used for amyloidosis are echocardiogram, magnetic resonance imaging (MRI), computed tomography (CT) scan, and nuclear imaging.
Biopsy: This is a procedure that involves taking a small sample of tissue from an affected organ or tissue, and examining it under a microscope for the presence of amyloid deposits. Biopsy is the definitive test to confirm the diagnosis of amyloidosis, and to identify the type of amyloid protein involved. A biopsy can be done from various sites, such as the fat under the skin, the bone marrow, the kidney, the liver, or the heart.
Laboratory tests: These are tests that analyze the blood and urine samples of the patient for abnormal levels of proteins, antibodies, or other substances that can indicate amyloidosis. Laboratory tests can also help to rule out other diseases that can cause similar symptoms and to monitor kidney and liver function, and blood counts. Some of the common laboratory tests used for amyloidosis are serum protein electrophoresis, urine protein electrophoresis, serum free light chain assay, serum amyloid A, and serum transthyretin.
Subtypes of Amyloid Proteins
Amyloid proteins are abnormal proteins that can form insoluble fibrils and deposit in various organs and tissues, causing amyloidosis. There are many different types of amyloid proteins, each with a different origin and structure. Some of the common subtypes of amyloid proteins are:
Amyloid beta (Aβ): This is a peptide of 36 to 43 amino acids that is derived from the amyloid precursor protein (APP), which is a transmembrane protein involved in neuronal development and function. Aβ is normally cleaved and degraded by enzymes, but in some cases, it can accumulate and form amyloid plaques in the brain. Aβ is the main component of the amyloid deposits found in Alzheimer’s disease, a neurodegenerative disorder that causes cognitive impairment and dementia. Aβ can also be found in other conditions, such as cerebral amyloid angiopathy, which is a disease that affects the blood vessels in the brain.
Transthyretin (TTR): This is a protein of 127 amino acids that is produced by the liver and the choroid plexus, which is a structure in the brain that produces cerebrospinal fluid. TTR is a carrier protein that transports thyroid hormones and vitamin A in the blood and the cerebrospinal fluid. TTR can undergo a conformational change and form amyloid fibrils, which can deposit in various organs and tissues. TTR is the main component of the amyloid deposits found in hereditary or familial amyloidosis, which is a genetic disorder that affects the nerves, heart, kidneys, and eyes. TTR can also be found in other conditions, such as senile systemic amyloidosis, which is a disease that affects the elderly and mainly involves the heart.
Immunoglobulin light chains (AL amyloidosis): These are fragments of antibodies that are produced by the plasma cells, which are a type of white blood cells that secrete antibodies to fight infections. Immunoglobulin light chains are normally paired with immunoglobulin heavy chains to form complete antibodies, but in some cases, they can be produced in excess and form amyloid fibrils, which can deposit in various organs and tissues. Immunoglobulin light chains are the main component of the amyloid deposits found in AL amyloidosis, which is also called primary amyloidosis. AL amyloidosis is usually associated with plasma cell dyscrasia, which is a disorder that causes abnormal growth and function of plasma cells. AL amyloidosis can affect the heart, kidneys, liver, and nerves.
Risk Factors for Amyloidosis
Amyloidosis is a rare disease that occurs when amyloid proteins build up in various organs and tissues of the body. The risk factors for amyloidosis may vary depending on the type and cause of the disease. Some of the common risk factors for amyloidosis are:
Age: The risk of amyloidosis increases with age, as the production and clearance of amyloid proteins may decline or become impaired over time. For example, the risk of Alzheimer’s disease, which is caused by Aβ amyloidosis, increases exponentially after the age of 65. The risk of senile systemic amyloidosis, which is caused by TTR amyloidosis, also increases with age, especially in men over 70. The risk of AL amyloidosis, which is caused by immunoglobulin light chain amyloidosis, is higher in people older than 40.
Genetics: Some types of amyloidosis are inherited, meaning that they are caused by mutations in genes that encode for amyloid proteins or enzymes that regulate them. For example, hereditary or familial amyloidosis is caused by mutations in the TTR gene, which result in abnormal forms of TTR that are more prone to forming amyloid fibrils. Other types of amyloidosis may have a genetic predisposition, meaning that they are influenced by genetic factors that increase the susceptibility or severity of the disease. For example, some variants of the APOE gene, which encodes for a protein that binds to Aβ, may increase the risk or accelerate the progression of Alzheimer’s disease.
Chronic diseases: Some types of amyloidosis are secondary, meaning that they are triggered or exacerbated by other diseases or conditions that cause chronic inflammation or infection. For example, AA amyloidosis is caused by the accumulation of serum amyloid A (SAA), which is a protein that is produced by the liver in response to inflammation or infection. AA amyloidosis can occur in people with chronic inflammatory diseases, such as rheumatoid arthritis, inflammatory bowel disease, or tuberculosis. AL amyloidosis can also be secondary to other diseases that cause abnormal production of immunoglobulin light chains, such as multiple myeloma, a type of blood cancer that affects plasma cells.
Complications and Impact on Organs
Amyloidosis can cause serious complications and impact the function and structure of various organs and tissues. The complications and impact of amyloidosis may depend on the type, location, and extent of the amyloid deposits. Some of the common complications and impacts of amyloidosis are:
Cardiac involvement: Amyloidosis can affect the heart, causing cardiomyopathy, which is a disease that affects the heart muscle. Cardiomyopathy can lead to heart failure, which is a condition that occurs when the heart cannot pump enough blood to meet the body’s needs. Heart failure can cause symptoms such as shortness of breath, swelling of the legs, fatigue, and irregular heartbeat. Amyloidosis can also affect the electrical system of the heart, causing arrhythmia, which is a disorder that affects the heart rhythm. Arrhythmia can cause symptoms such as palpitations, dizziness, fainting, and sudden cardiac death.
Renal dysfunction: Amyloidosis can affect the kidneys, causing nephropathy, which is a disease that affects kidney function. Nephropathy can lead to kidney failure, which is a condition that occurs when the kidneys cannot filter waste and excess fluid from the blood. Kidney failure can cause symptoms such as protein in the urine, swelling of the body, high blood pressure, anemia, and bone disease. Kidney failure may require dialysis, which is a treatment that uses a machine to remove waste and fluid from the blood, or kidney transplant, which is a surgery that replaces a damaged kidney with a healthy one from a donor.
Neurological complications: Amyloidosis can affect the nervous system, causing neuropathy, which is a disease that affects the nerves. Neuropathy can cause symptoms such as numbness, tingling, pain, or weakness in the hands, feet, or other parts of the body. Neuropathy can also affect the autonomic nervous system, which controls involuntary functions such as blood pressure, heart rate, digestion, and sweating. Autonomic neuropathy can cause symptoms such as dizziness, fainting, nausea, vomiting, diarrhea, constipation, erectile dysfunction, and urinary problems. Amyloidosis can also affect the brain, causing dementia, which is a decline in cognitive abilities such as memory, thinking, and reasoning. Dementia can cause symptoms such as confusion, disorientation, personality changes, and behavioral problems.
Treatment Approaches
The treatment of amyloidosis depends on the type, cause, and extent of the disease. There is no cure for amyloidosis, but treatments can help manage the symptoms, slow down the progression, and improve the quality of life of people with the condition. Some of the treatment approaches for amyloidosis are:
Targeting underlying causes: This approach aims to reduce or eliminate the source of the amyloid protein that causes the disease. For example, in AL amyloidosis, which is caused by abnormal antibodies produced by plasma cells, treatments such as chemotherapy, stem cell transplant, or targeted therapy can suppress or destroy the plasma cells and reduce the production of amyloid. In AA amyloidosis, which is caused by chronic inflammation or infection, treatments such as anti-inflammatory drugs, antibiotics, or biologics can control or cure the underlying condition and reduce the production of amyloid. In hereditary or familial amyloidosis, which is caused by genetic mutations, treatments such as gene therapy, liver transplant, or RNA interference can correct or silence the faulty gene and prevent the production of amyloid.
Symptomatic relief and supportive care: This approach aims to alleviate the symptoms and complications of amyloidosis and prevent further organ damage. For example, in cardiac amyloidosis, which affects the heart, treatments such as diuretics, beta-blockers, anticoagulants, or pacemakers can help reduce fluid retention, control heart rate, prevent blood clots, or regulate heart rhythm. In renal amyloidosis, which affects the kidneys, treatments such as dialysis or kidney transplant can help remove waste and fluid from the blood and restore kidney function. In neurological amyloidosis, which affects the nerves, treatments such as painkillers, antidepressants, anticonvulsants, or nerve stimulators can help relieve pain, depression, seizures, or nerve damage.
Emerging therapies and research: This approach aims to develop new and better treatments for amyloidosis that can target the amyloid deposits directly and dissolve them or prevent them from forming. For example, some emerging therapies include monoclonal antibodies, which are proteins that can bind to and eliminate the amyloid; chaperone molecules, which are substances that can stabilize the normal shape of the amyloid protein and prevent it from misfolding; and immunotherapy, which is a treatment that can stimulate the immune system to attack and destroy the amyloid.
Prognosis and Outlook
Amyloidosis is a rare and serious disease that occurs when abnormal proteins called amyloids accumulate in various organs and tissues of the body. The prognosis and outlook of amyloidosis depend on several factors, such as:
The type of amyloidosis: There are many different types of amyloidosis, each with a different cause and a different type of amyloid protein. Some types of amyloidosis are inherited, while others are acquired. Some types of amyloidosis are systemic, affecting multiple organs and tissues, while others are localized, affecting only one organ or tissue. The type of amyloidosis determines the treatment options, the response to treatment, and the risk of complications and organ failure.
The severity of organ involvement: Amyloidosis can affect any organ or tissue in the body, but the most common and serious ones are the heart, kidneys, liver, nerves, and digestive system. The extent and degree of organ damage caused by amyloid deposits can vary from person to person, and can affect the symptoms, the quality of life, and the survival of people with amyloidosis. The prognosis and outlook of amyloidosis are often worse when the heart is involved, as cardiac amyloidosis can lead to heart failure, arrhythmia, and sudden death
The response to treatment: The treatment of amyloidosis aims to reduce the production or remove the source of amyloid proteins, and to manage the symptoms and complications of organ dysfunction. The treatment options vary depending on the type and cause of amyloidosis and may include chemotherapy, immunotherapy, targeted therapy, organ transplantation, or supportive care. The response to treatment can be measured by the reduction of amyloid proteins in the blood or urine, the improvement of organ function, and the resolution of symptoms. The response to treatment can affect the prognosis and outlook of amyloidosis, as a better response can lead to longer survival and better quality of life
Despite the challenges and uncertainties of amyloidosis, there have been significant advances in the diagnosis, treatment, and management of this disease in recent years. These advances include:
The development of new diagnostic tools: The diagnosis of amyloidosis can be difficult and delayed, as the symptoms can be nonspecific and similar to those of other diseases. However, new diagnostic tools have been developed or improved to help with the detection and identification of amyloidosis. These tools include imaging techniques, such as cardiac magnetic resonance imaging (MRI), nuclear scintigraphy, and positron emission tomography (PET), which can visualize the amyloid deposits in the organs and tissues.These tools also include laboratory tests, such as serum free light chain assay, serum amyloid A, and serum transthyretin, which can measure the levels of amyloid proteins in the blood. These tools also include biopsy methods, such as laser microdissection and mass spectrometry, which can analyze the type and composition of amyloid proteins in the tissue samples.
The discovery of new treatment options: The treatment of amyloidosis has been evolving and expanding, as new treatment options have been discovered or approved for different types of amyloidosis. These options include novel drugs, such as daratumumab, ixazomib, and venetoclax, which can target the underlying plasma cell dyscrasia or B-cell neoplasm that produces the amyloid proteins. These options also include gene therapies, such as patisiran and inotersen, which can silence the expression of the mutated transthyretin gene that causes hereditary amyloidosis. These options also include immunotherapies, such as NEOD001 and CAEL-101, which can bind to and clear the amyloid deposits from the organs and tissues.
The improvement of supportive care: The supportive care of amyloidosis aims to alleviate the symptoms and complications of organ dysfunction, and to improve the quality of life and survival of people with amyloidosis. The supportive care of amyloidosis has been improved by the optimization of the dosing and timing of the medications, the prevention and management of infections and bleeding, and the provision of palliative and hospice care.
However, amyloidosis remains a challenging and complex disease that requires timely diagnosis and intervention, as well as multidisciplinary and individualized care. Therefore, it is important to raise awareness and research on amyloidosis, to enhance the understanding and recognition of this disease, to develop more effective and safer treatment options, and to improve the outcomes and quality of life of people with amyloidosis. 😊
FAQ
01. What are the 4 types of amyloidosis?
There are many different types of amyloidosis, but the four main types are:
AL amyloidosis: This is the most common type of amyloidosis, which is caused by the accumulation of immunoglobulin light chains, which are fragments of antibodies produced by abnormal plasma cells. It usually affects the heart, kidneys, liver, and nerves.
AA amyloidosis: This is a rare type of amyloidosis, which is caused by the accumulation of serum amyloid A (SAA), which is a protein produced by the liver in response to chronic inflammation or infection. It usually affects the kidneys, spleen, liver, and intestines.
ATTR amyloidosis: This is a type of hereditary or familial amyloidosis, which is caused by mutations in the transthyretin (TTR) gene, which encodes for a protein that transports thyroid hormones and vitamin A. It usually affects the nerves, heart, kidneys, and eyes.
Aβ amyloidosis: This is a type of amyloidosis that affects the brain, causing Alzheimer’s disease, which is a neurodegenerative disorder that causes cognitive impairment and dementia. It is caused by the accumulation of amyloid beta (Aβ), which is a peptide derived from the amyloid precursor protein (APP).
02. What is the main cause of amyloidosis?
The main cause of amyloidosis is the abnormal accumulation of amyloid proteins in various organs and tissues of the body. Amyloid proteins are misfolded or unstable proteins that can form insoluble fibrils and deposit in the extracellular space, interfering with the normal function and structure of the organs and tissues. The cause of amyloidosis may vary depending on the type and source of the amyloid protein. Some amyloid proteins are produced by the bone marrow or by certain cells in response to inflammation or infection, while others are inherited or acquired due to mutations or alterations in the genes that encode for them.
03. What are the three major types of amyloidosis?
The three major types of amyloidosis are:
Systemic amyloidosis: This is the type of amyloidosis that affects multiple organs and tissues throughout the body. Systemic amyloidosis can be further classified into subtypes based on the source and nature of the amyloid protein, such as AL, AA, and ATTR amyloidosis.
Localized amyloidosis: This is the type of amyloidosis that affects only one organ or tissue. Localized amyloidosis can be caused by overproduction of amyloid by the cells in that specific area, or by deposition of amyloid from the blood. Localized amyloidosis can affect the skin, eyes, lungs, brain, or other parts of the body.
Primary and secondary amyloidosis: These are terms that are sometimes used to describe the cause of amyloidosis. Primary amyloidosis refers to amyloidosis that occurs without any known underlying disease or trigger, such as AL amyloidosis. Secondary amyloidosis refers to amyloidosis that occurs as a complication of another disease or condition, such as chronic infection, inflammation, or cancer, such as AA amyloidosis.
04. Is there any treatment for amyloidosis?
Yes, there are various treatment options for amyloidosis, depending on the type, cause, and extent of the disease. The treatment of amyloidosis aims to reduce the production or remove the source of amyloid proteins, and to manage the symptoms and complications of organ dysfunction. The treatment options may include:
Chemotherapy: This is a treatment that uses drugs to kill or stop the growth of abnormal cells that produce amyloid proteins, such as plasma cells or B cells. Chemotherapy is often used for AL amyloidosis, which is caused by immunoglobulin light-chain amyloidosis. Chemotherapy may also be used for other types of amyloidosis that are associated with blood cancers, such as multiple myeloma or lymphoma.
Immunotherapy: This is a treatment that uses drugs or substances that stimulate or modulate the immune system to fight the amyloid proteins or the abnormal cells that produce them. Immunotherapy may include monoclonal antibodies, which are proteins that bind to specific targets on the amyloid proteins or the abnormal cells, or immunomodulators, which are drugs that enhance or suppress the immune response. Immunotherapy may be used for AL amyloidosis or other types of amyloidosis that involve the immune system.
Targeted therapy: This is a treatment that uses drugs that target specific molecules or pathways that are involved in the formation or deposition of amyloid proteins. Targeted therapy may include proteasome inhibitors, which are drugs that block the enzyme that breaks down the amyloid proteins, or kinase inhibitors, which are drugs that block the enzyme that activates the abnormal cells that produce the amyloid proteins. Targeted therapy may be used for AL amyloidosis or other types of amyloidosis that involve specific molecular mechanisms.
Gene therapy: This is a treatment that uses genetic engineering to modify or replace the genes that encode for the amyloid proteins or the enzymes that regulate them. Gene therapy may include RNA interference, which is a technique that uses small molecules of RNA to silence the expression of the mutated or overexpressed genes, or gene editing, which is a technique that uses molecular tools to correct or insert the normal or modified genes. Gene therapy may be used for hereditary or familial amyloidosis, such as ATTR amyloidosis, which is caused by mutations in the TTR gene.
Organ transplantation: This is a treatment that involves replacing a damaged organ with a healthy one from a donor. Organ transplantation may be used for amyloidosis that causes severe or irreversible organ failure, such as cardiac or renal amyloidosis. Organ transplantation may also be used for amyloidosis that is resistant or refractory to other treatments, or that recurs after other treatments. Organ transplantation may require immunosuppressive drugs to prevent the rejection of the donor organ by the recipient’s immune system.
Supportive care: This is a treatment that aims to alleviate the symptoms and complications of organ dysfunction, and to improve the quality of life and survival of people with amyloidosis. Supportive care may include medications, such as diuretics, beta-blockers, or anticoagulants, to manage the heart failure, arrhythmia, or blood clots caused by cardiac amyloidosis, or dialysis, which is a treatment that uses a machine to remove waste and fluid from the blood, to manage the kidney failure caused by renal amyloidosis. Supportive care may also include dietary modifications, fluid restriction, or nutritional supplements, to manage the weight loss, malnutrition, or diarrhea caused by digestive amyloidosis. Supportive care may also include pain relief, physical therapy, or nerve stimulation, to manage the numbness, tingling, pain, or weakness caused by neurological amyloidosis. Supportive care may also include palliative or hospice care, which are services that provide comfort and support to people with terminal or incurable diseases.
05. What is the best treatment for amyloidosis?
There is no definitive answer to what is the best treatment for amyloidosis, as different treatments have different advantages and disadvantages in terms of their effectiveness, safety, and availability. The best treatment for amyloidosis depends on several factors, such as:
The type and cause of amyloidosis: Different types of amyloidosis require different treatments, as they have different sources and natures of the amyloid proteins. For example, AL amyloidosis, which is caused by immunoglobulin light chain amyloidosis, may require chemotherapy, immunotherapy, or targeted therapy, while ATTR amyloidosis, which is caused by mutations in the TTR gene, may require gene therapy or organ transplantation.
The severity and location of organ involvement: The extent and degree of organ damage caused by amyloid deposits can affect the choice and response of treatment, as some treatments may be more effective or safer for certain organs or tissues than others. For example, cardiac amyloidosis, which affects the heart, may require organ transplantation or supportive care, while renal amyloidosis, which affects the kidneys, may require dialysis or supportive care.
The patient’s condition and preferences: The patient’s age, weight, kidney function, and other medical conditions can affect the tolerance and outcome of treatment, as some treatments may have more side effects or complications than others. For example, chemotherapy, immunotherapy, or targeted therapy may cause nausea, vomiting, hair loss, or infections, while gene therapy or organ transplantation may cause allergic reactions, bleeding, or rejection. The patient’s preferences, values, and goals can also affect the decision and consent of treatment, as some treatments may have more benefits or risks than others. For example, some patients may prefer a more aggressive or invasive treatment, while others may prefer a more conservative or palliative treatment.
Therefore, the best treatment for amyloidosis is the one that is most suitable and effective for the individual patient, based on the type, cause, and extent of the disease, and the patient’s condition and preferences. The best treatment for amyloidosis should be determined by a multidisciplinary team of specialists, such as hematologists, cardiologists, nephrologists, neurologists, gastroenterologists, and geneticists, in consultation with the patient and their family or caregivers.
06. What is the early treatment of amyloidosis?
The early treatment of amyloidosis is crucial to prevent or delay the progression and complications of the disease. The early treatment of amyloidosis depends on the type, cause, and extent of the disease, and may include:
Targeting the underlying cause: This is the main goal of the early treatment of amyloidosis, as it can reduce the production or remove the source of the amyloid proteins, and prevent further organ damage. The treatment options may vary depending on the cause of amyloidosis, such as chemotherapy, immunotherapy, targeted therapy, gene therapy, or organ transplantation. The early treatment of the underlying cause can improve the survival and quality of life of people with amyloidosis.
Symptomatic relief and supportive care: This is the secondary goal of the early treatment of amyloidosis, as it can alleviate the symptoms and complications of organ dysfunction, and improve the quality of life and survival of people with amyloidosis. The treatment options may include medications, such as diuretics, beta-blockers, or anticoagulants, to manage heart failure, arrhythmia, or blood clots caused by cardiac amyloidosis, or dialysis, which is a treatment that uses a machine to remove waste and fluid from the blood, to manage the kidney failure caused by renal amyloidosis. The treatment options may also include dietary modifications, fluid restriction, or nutritional supplements, to manage the weight loss, malnutrition, or diarrhea caused by digestive amyloidosis. The treatment options may also include pain relief, physical therapy, or nerve stimulation, to manage the numbness, tingling, pain, or weakness caused by neurological amyloidosis. The treatment options may also include palliative or hospice care, which are services that provide comfort and support to people with terminal or incurable diseases.
07. How treatable is amyloidosis?
Amyloidosis is a treatable disease, but the effectiveness and safety of the treatment may vary depending on the type, cause, and extent of the disease, and the patient’s condition and preferences. The treatment of amyloidosis aims to reduce the production or remove the source of amyloid proteins, and to manage the symptoms and complications of organ dysfunction. The treatment options may include chemotherapy, immunotherapy, targeted therapy, gene therapy, organ transplantation, or supportive care. The response to treatment can be measured by the reduction of amyloid proteins in the blood or urine, the improvement of organ function, and the resolution of symptoms. The response to treatment can affect the prognosis and outlook of amyloidosis, as a better response can lead to longer survival and better quality of life.
08. Do people recover from amyloidosis?
Some people may recover from amyloidosis, while others may not. The recovery from amyloidosis depends on several factors, such as:
The type and cause of amyloidosis: Some types of amyloidosis are more likely to be cured or controlled than others, depending on the source and nature of the amyloid proteins. For example, AL amyloidosis, which is caused by immunoglobulin light chain amyloidosis, may be cured or controlled by chemotherapy, immunotherapy, or targeted therapy, while ATTR amyloidosis, which is caused by mutations in the TTR gene, may be cured or controlled by gene therapy or organ transplantation.
The severity and location of organ involvement: The extent and degree of organ damage caused by amyloid deposits can affect the recovery from amyloidosis, as some organs or tissues may be more resilient or regenerative than others. For example, the kidneys may recover from amyloidosis if the amyloid deposits are removed or reduced, while the heart may not recover from amyloidosis if the amyloid deposits cause irreversible fibrosis or scarring.
The response to treatment: The recovery from amyloidosis depends on the effectiveness and safety of the treatment, as well as the patient’s compliance and adherence to the treatment. The recovery from amyloidosis can be assessed by the reduction of amyloid proteins in the blood or urine, the improvement of organ function, and the resolution of symptoms. The recovery from amyloidosis can also be influenced by the side effects or complications of the treatment, such as infections, bleeding, or rejection.
09. Can amyloidosis go away?
Amyloidosis can go away in some cases, but not in others. The possibility of amyloidosis going away depends on several factors, such as:
The type and cause of amyloidosis: Some types of amyloidosis are more likely to go away than others, depending on the source and nature of the amyloid proteins. For example, AA amyloidosis, which is caused by chronic inflammation or infection, may go away if the underlying condition is treated or resolved, while AL amyloidosis, which is caused by immunoglobulin light chain amyloidosis, may not go away unless the abnormal plasma cells are eradicated or suppressed.
The severity and location of organ involvement: The extent and degree of organ damage caused by amyloid deposits can affect the possibility of amyloidosis going away, as some organs or tissues may be more prone or resistant to amyloid clearance than others. For example, the skin may go away from amyloidosis if the amyloid deposits are removed by surgery or laser, while the brain may not go away from amyloidosis if the amyloid deposits are inaccessible or irreversible.
The response to treatment: The possibility of amyloidosis going away depends on the effectiveness and safety of the treatment, as well as the patient’s compliance and adherence to the treatment. The possibility of amyloidosis going away can be assessed by the reduction or disappearance of amyloid proteins in the blood or urine, the normalization or restoration of organ function, and the absence or improvement of symptoms. The possibility of amyloidosis going away can also be influenced by the side effects or complications of the treatment, such as infections, bleeding, or rejection.
10. Can you live a long life with amyloidosis?
Some people can live a long life with amyloidosis, while others may not. The life expectancy of people with amyloidosis depends on several factors, such as:
The type and cause of amyloidosis: Some types of amyloidosis have a better or worse prognosis than others, depending on the source and nature of the amyloid proteins. For example, AL amyloidosis, which is caused by immunoglobulin light chain amyloidosis, has a poor prognosis, with a median survival of 1 to 2 years, while ATTR amyloidosis, which is caused by mutations in the TTR gene, has a variable prognosis, with a median survival of 5 to 15 years.
The severity and location of organ involvement: The extent and degree of organ damage caused by amyloid deposits can affect the life expectancy of people with amyloidosis, as some organs or tissues are more vital or vulnerable than others. For example, cardiac amyloidosis, which affects the heart, has a worse prognosis than renal amyloidosis, which affects the kidneys, as the heart is more essential and sensitive to amyloid deposition.
The response to treatment: The life expectancy of people with amyloidosis depends on the effectiveness and safety of the treatment, as well as the patient’s compliance and adherence to the treatment. The life expectancy of people with amyloidosis can be improved by the reduction or elimination of amyloid proteins in the blood or urine, the improvement or preservation of organ function, and the relief or prevention of symptoms and complications. The life expectancy of people with amyloidosis can also be reduced by the side effects or complications of the treatment, such as infections, bleeding, or rejection.
11. What is end stage amyloidosis?
End-stage amyloidosis is the final stage of amyloidosis, which occurs when the amyloid deposits cause severe and irreversible organ failure, and the treatment options are exhausted or ineffective. End-stage amyloidosis can affect any organ or tissue in the body, but the most common and serious ones are the heart, kidneys, liver, nerves, and digestive system. End-stage amyloidosis can cause symptoms such as shortness of breath, chest pain, irregular heartbeat, fluid retention, protein in the urine, swelling of the legs, high blood pressure, kidney failure, jaundice, liver failure, numbness, tingling, pain, weakness, diarrhea, constipation, nausea, vomiting, weight loss, and malnutrition. End-stage amyloidosis can also cause complications such as bleeding, infections, and blood clots. End-stage amyloidosis can be fatal, and the survival rate is very low.
12. Is amyloidosis permanent?
Amyloidosis can be permanent in some cases, but not in others. The permanence of amyloidosis depends on several factors, such as:
The type and cause of amyloidosis: Some types of amyloidosis are more likely to be permanent than others, depending on the source and nature of the amyloid proteins. For example, AL amyloidosis, which is caused by immunoglobulin light chain amyloidosis, may be permanent unless the abnormal plasma cells are eradicated or suppressed, while AA amyloidosis, which is caused by chronic inflammation or infection, may not be permanent if the underlying condition is treated or resolved.
The severity and location of organ involvement: The extent and degree of organ damage caused by amyloid deposits can affect the permanence of amyloidosis, as some organs or tissues may be more resilient or regenerative than others. For example, the kidneys may not be permanent from amyloidosis if the amyloid deposits are removed or reduced, while the heart may be permanent from amyloidosis if the amyloid deposits cause irreversible fibrosis or scarring.
The response to treatment: The permanence of amyloidosis depends on the effectiveness and safety of the treatment, as well as the patient’s compliance and adherence to the treatment. The permanence of amyloidosis can be reversed by the reduction or elimination of amyloid proteins in the blood or urine, the improvement or restoration of organ function, and the resolution of symptoms. The permanence of amyloidosis can also be maintained by the side effects or complications of the treatment, such as infections, bleeding, or rejection.
13. Is amyloidosis harmful?
Amyloidosis is a harmful disease, as it can cause serious and potentially life-threatening complications and impact the function and structure of various organs and tissues. Amyloidosis can affect any organ or tissue in the body, but the most common and serious ones are the heart, kidneys, liver, nerves, and digestive system. Amyloidosis can cause symptoms such as shortness of breath, chest pain, irregular heartbeat, fluid retention, protein in the urine, swelling of the legs, high blood pressure, kidney failure, jaundice, liver failure, numbness, tingling, pain, weakness, diarrhea, constipation, nausea, vomiting, weight loss, and malnutrition. Amyloidosis can also cause complications such as bleeding, infections, and blood clots. Amyloidosis can be fatal, and the survival rate is very low.
*Image credits- freepik*
Important Notice:
The information provided on “health life ai” is intended for informational purposes only. While we have made efforts to ensure the accuracy and authenticity of the information presented, we cannot guarantee its absolute correctness or completeness. Before applying any of the strategies or tips, please consult a professional medical adviser.